ICHTHYOSIS
• Ichthyosis Vulgaris
• X-Linked Ichthyosis
• Epidermolytic Hyperkeratosis
• Autosomal Recessive Ichthyosis
Ichthyosis Vulgaris
• Mutations in filaggrin (AD)
• a few months after birth
• The flexural creases are spared
– Keratosis pilaris is often present, and
– The palms and soles may show hyperkeratosis.
• Acquired, noninherited form with lymphoma, particularly Hodgkin's disease
– also with carcinoma and sarcoidosis.
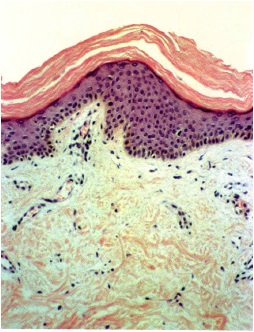
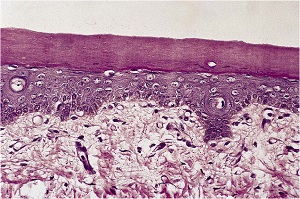
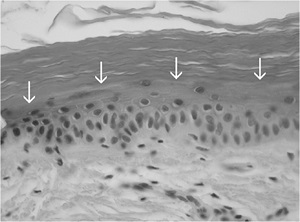
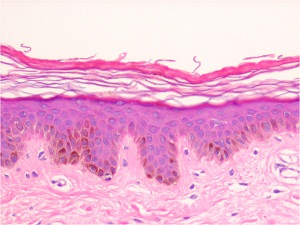
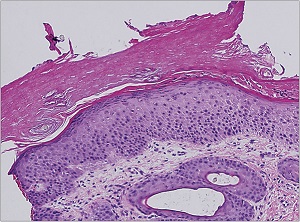
Histopathology
• A moderate degree of hyperkeratosis with a thin or absent granular layer
• The hyperkeratosis often extends into the hair follicles, resulting in large keratotic follicular plugs.
• The dermis is normal.
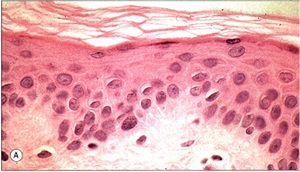
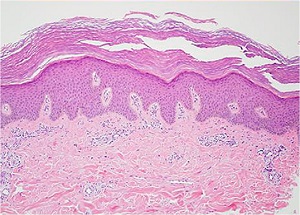
X-Linked Ichthyosis
• 90% caused by gene deletion of the steroid sulfatase gene
• Rarely present at birth, scales increases during childhood
• The flexural creases may be involved
• Delayed dissolution of the desmosomal disks in the horny layer
Histopathology
• Slightly thickened epidermis with hyperkeratosis and normal or slightly thickened granular layer
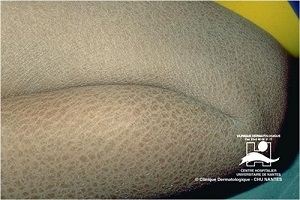
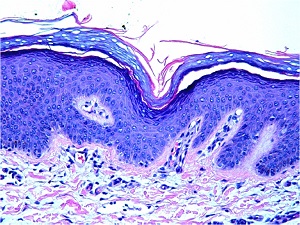
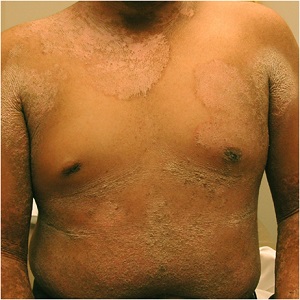
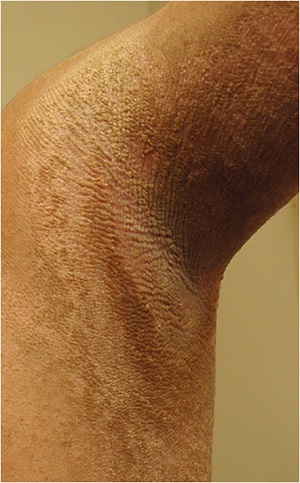
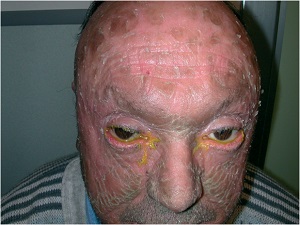
Epidermolytic Hyperkeratosis
(bullous congenital ichthyosiform erythroderma)
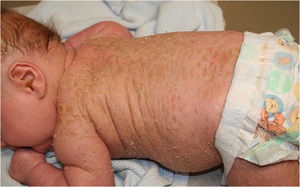
• Generalized erythema from the time of birth
• Flexural surfaces of the extremities show marked involvement
• Pathogenesis. Defects in keratin genes (KRT1 and KRT10)
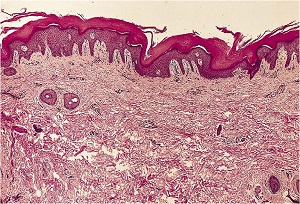
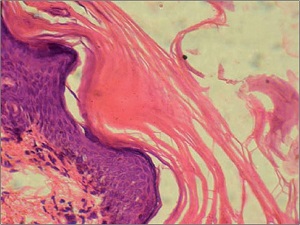
Histopathology
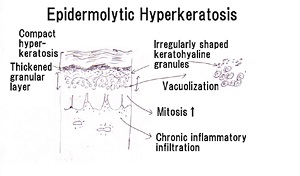
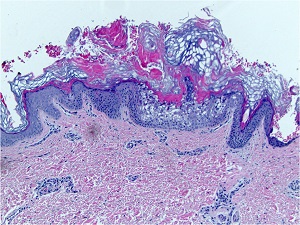
• Characteristic histologic picture is epidermoytic hyperkeratosis ; vacuolar and granular degeneration in mid and upper epidermis
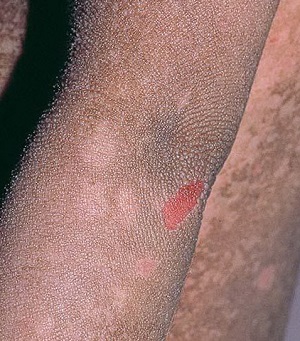
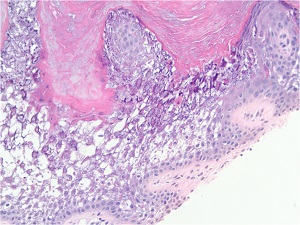
– in bullous as well as in nonbullous areas
• Variously sized clear spaces around the nuclei and peripheral to the clear spaces, the cells show indistinct boundaries formed by lightly staining material or by keratohyaline granules.
• Granular layer is thickened and contains irregularly shaped keratohyaline granules and is covered by compact hyperkeratosis
• Upper dermis shows a moderately severe, chronic inflammatory infiltrate.
• Mitotic figures are five times more numerous than in normal epidermis
Autosomal Recessive Ichthyosis
• A less severe type, congenital ichthyosiform erythroderma,
– fine white scales with fairly pronounced erythroderma
– tendency to improve at puberty
• The more severe type, lamellar ichthyosis,
– large, plate like scales and severe ectropion
– slight erythroderma
• In both forms, the flexural surfaces and the palms and soles are involved
Histopathology
• The histologic findings in both congenital ichthyosiform erythroderma and lamellar ichthyosis are nonspecific.
• As a rule
– congenital ichthyosiform erythroderma:
• mild hyperkeratosis with foci of parakeratosis
– lamellar ichthyosis
• Marked hyperkeratosis without parakeratosis
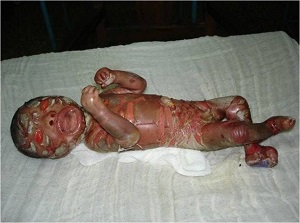
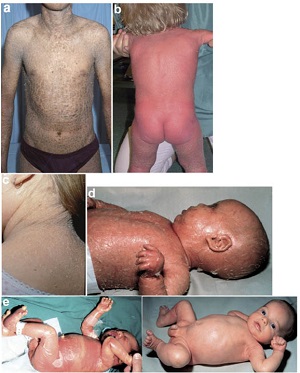
• a) lamellar ichthyosis (LI), b) congenital ichthyosiform erythroderma (CIE) or erythrodermic ichtyosis (EI), c) congenital ichthyosis with fine or focal scaling (CIFS), d) Harelquin ichthyosis (HI)
• e) self-healing collodion baby (SHCB) soon after birth (left) and at 2 month of age (right).
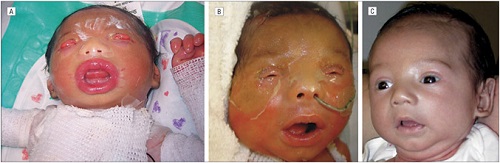
• Self-Healing Collodion Membrane and Mild Nonbullous Congenital Ichthyosiform Erythroderma
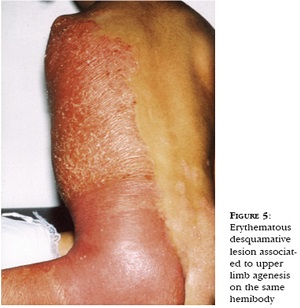
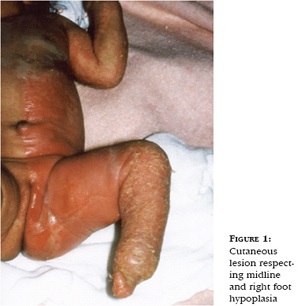
CHILD Syndrome
Congenital Hemidysplasia with Ichthyosiform
erythrodermal and Limb Defects
• From birth unilateral ichthyosiform erythroderma and ipsilateral underdevelopment of the limbs
• X-linked dominant that is lethal in the hemizygote male fetus
• Mutations in genes encoding enzymes involved in sequential steps in the conversion of lanosterol to cholesterol
• Histopathology:
– thickened epidermis,
– pronounce hyperkeratosis with prominent parakeratotic foci
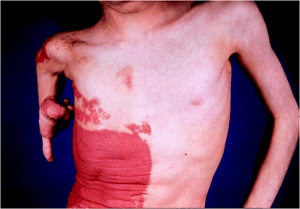
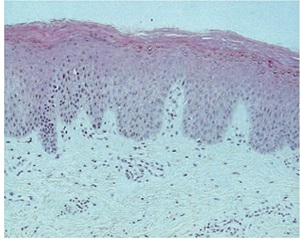
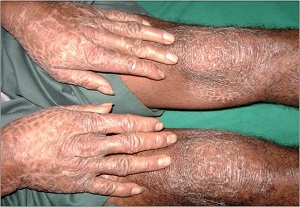
Erythrokeratodermia Variabilis
• Rare, AD, starts in infancy rather than at birth.
• Two morpho-logic components.
– Areas of variable erythema that expand and coalesce into circinate
– Persistent hyperkeratotic plaques both within the areas of erythema and in areas of apparently normal skin
• In Progressive symmetric erythrokeratodermia
– only persistent erythematous hyperkeratotic plaques
– limited to the extremities
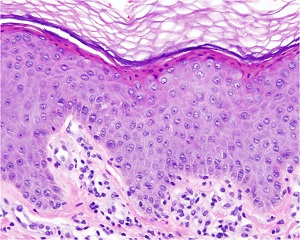
Histopathology
• Nonspecific
• In the hyperkeratotic plaques
– Hyperkeratosis with moderate papillomatosis and acanthosis. The granular layer appears normal
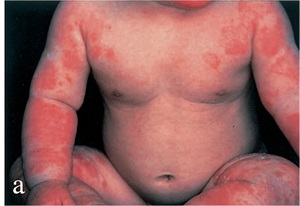
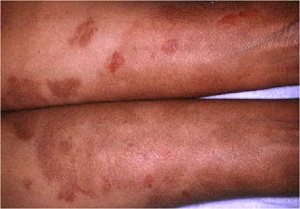
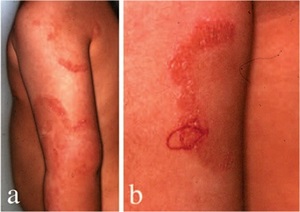
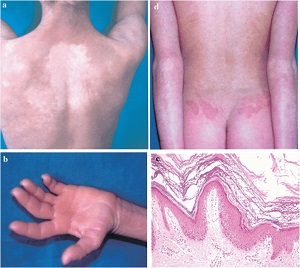
Ichthyosis Linearis Circumflexa
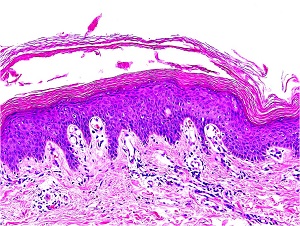
• AR, presents at birth or starts shortly thereafter
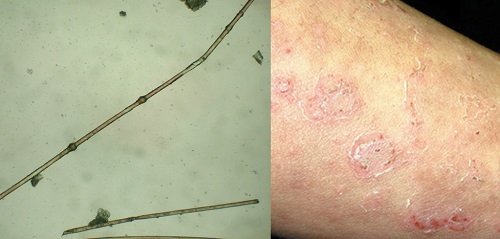
• Extensive migratory polycyclic lesions of erythema and,some of the areas show a distinctive "double edged“ scaling at their periphery
• Persists through life
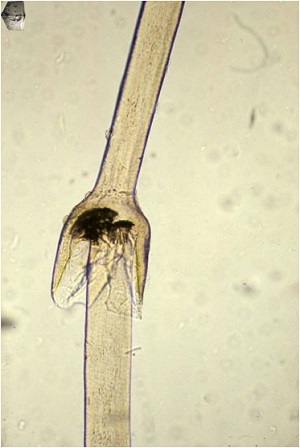
• More than half as part of Netherton's syndrome hair anomalies have been present in the scalp, usually trichorrhexis invaginata
Histopathology
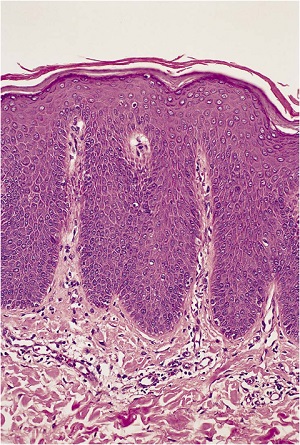
• The areas of erythema and scaling show nonspecific changes with some resemblance to psoriasis, such as
– elongation of the rete ridges
– Hyperkeratosis, as well as parakeratosis
• The double-edged scale frequently shows in the upper irregular spongiosis, or vesiculopustules within the horny layer
• Focal accumulations of PAS positive, diastase-resistant, homogeneous material are seen within a parakeratotic stratum corneum
• Netherton syndrome (NS) is caused by mutations in serine protease inhibitor Kazal-type 5 (SPINK5), which encodes the lymphoepithelial Kazal-type-related inhibitor (LEKTI)
• Lack of LEKTI causes stratum corneum detachment secondary to the hyperactivity of epidermal proteases, which leads to skin barrier defect, thus facilitating allergen penetration and contributing to atopy in NS
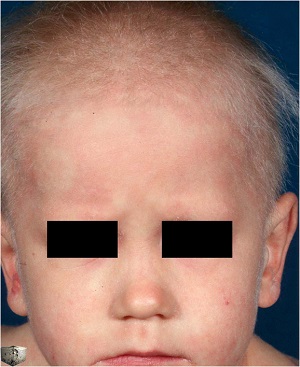
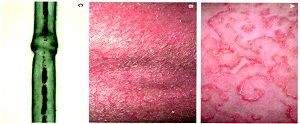
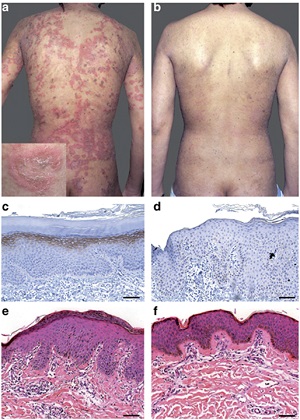
• Clinical and histological features of Netherton syndrome. (a) Clinical photograph of the back showing characteristic ichthyosis linearis circumflexa with polycyclic erythematous patches surrounded by a serpiginous overlying double-edged scale (enlargement). (b) Clinical evolution of skin lesions after 12 infusions of 5 mg kg−1 infliximab. Immunostaining of LEKTI in (c) normal human skin and (d) the patient's skin. Histopathological images from the patient's skin (e) before and (f) after 12 infusions of infliximab. Note the reduced inflammatory infiltrate and the absence of subcorneal pustule in panel f.
Syndromes Associated with Ichthyosis
• Sjogren-Larsson syndrome
– lamellar ichthyosis, mental retardation, spastic paresis
– Fatty alcohol and NAD oxidoreductase enzyme activity deficiency in skin fibroblasts and leukocytes
– a gene defect in FALDH
• Rud's syndrome
– Generalized ichthyosis, hypogonadism, mental deficiency, and epilepsy
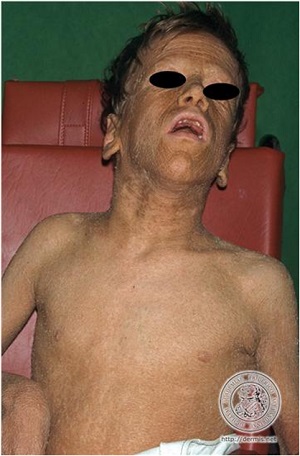
• Conradi-Hunermann syndrome
– X-linked
– chondrodysplasia punctata
– ichthyosis with a whorled pattern
– skeletal and ocular abnormalities
– gene defect in emopamil binding protein affecting delta 8, delta 7 sterol isomerase
– Some forms are associated with peroxisome abnormalities
• IBIDS syndrome
– icthyosis associated
– brittle hair: (sulfur-deficient, sparse hair)
– impaired intelligence, decreased fertility, and short stature
• PIBIDS syndrome
– photosensitivity and IBIDS.
• KID syndrome
– keratitis associated with
– ichthyosis and deafness
• neutral lipid storage disease,
– ichthyosis with cataracts, deafness, ataxia,
– lipid droplets in many circulating cells
• multiple sulfatase deficiencies,
– Ichthyosis,
– neurodegeneration, organomegaly,skeletal dysplasia
• The only syndrome showing specific histologic changes in the skin is Refsum's syndrom
Refsum's Syndrome
• also called
– Phytanic Acid Storage Disease
– Heredopathia Atactica Polyneuritiformis
• autosomal recessive disorder
• generalized ichthyosis, cerebellar ataxia, progressive paresis of the extremities, and retinitis pigmentosa.
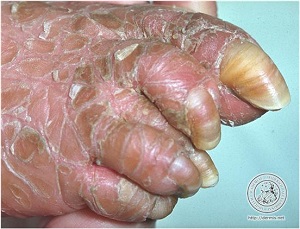
• Refsum's disease in an Arabian patient. Symmetric short fourth toes (arrow).
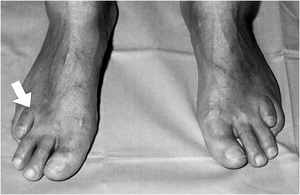
REFSUMS SYNDROME - RETINITIS PIGMENTOSA
Histopathology and pathogenesis
• hyperkeratosis, hypergranulosis, and acanthosis.
• In the basal and suprabasal cells of the epidermis are
– variably sized vacuoles that on staining for lipids are seen to contain lipid accumulations
• accumulation of phytanic acid
– deficiency of alpha-phytanic acid alpha-hydroxylase
– mutations in the phytanol-CoA hydroxylase gene
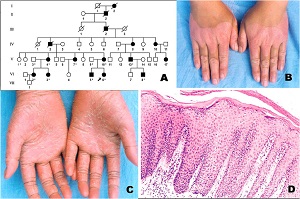
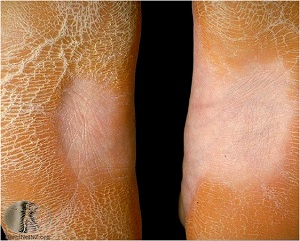
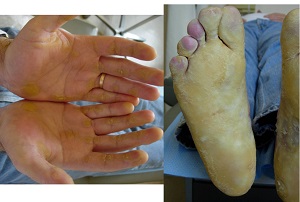
KERATOSIS PALMARIS ET PLANTARIS
• Three major autosomal dominant forms
– Unna- Thost
• diffuse or localized, occasionally linear
• an extending type with gradual progression to the extensors
– Epidermolytic
• Clinically indistinguishable from the Unna
– punctata (or papulosa)
• two autosomal recessive forms
– Meleda type
• Marked tendency toward progression to the extensors
– Papillon-Lefevre syndrome
• clinical characteristics of the Meleda type in association with
• periodontosis
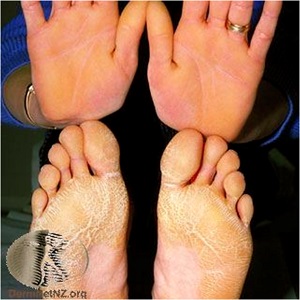
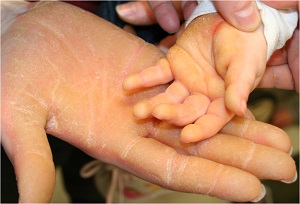
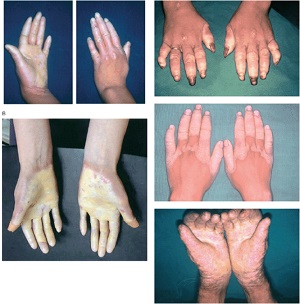
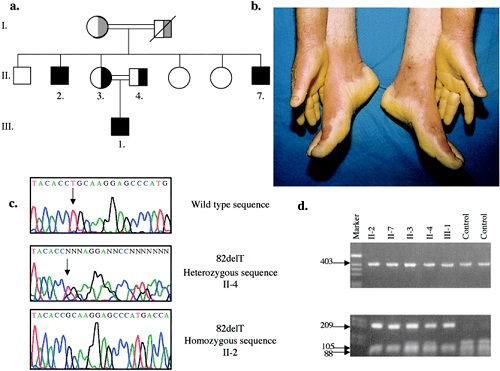
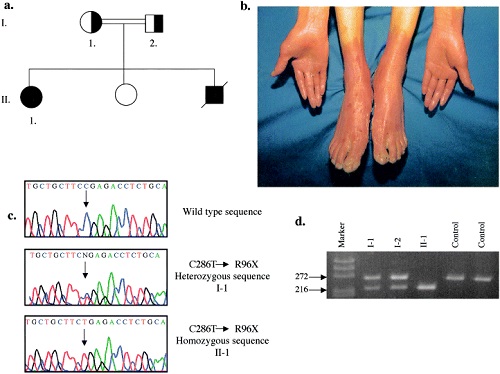
keratosis palmaris et plantaris in syndromes
• Pachyonychia congenita,
• Hidrotic ectodermal dysplasia,
• Richner-Hanhart syndrome associated with tyrosinemia (tyrosine tyrosinase on 16q22,I-q22.3)
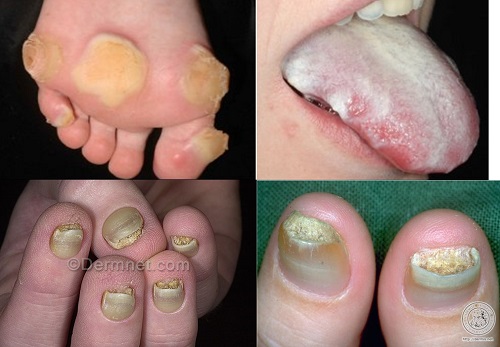
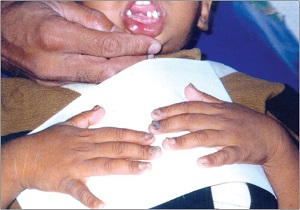
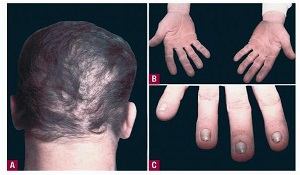
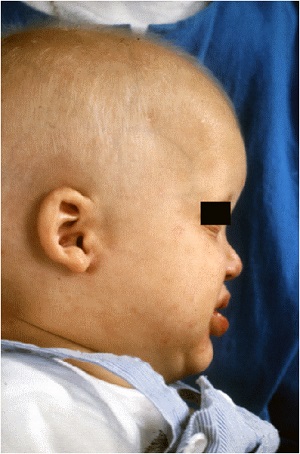
• autosomal dominant
• defective
– hair
– nails,
– keratoderma of the palms and soles
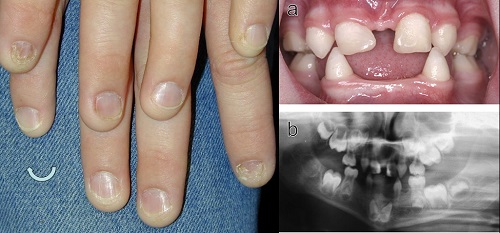
Tyrosinemia type II or Richner–Hanhart Syndrome (RHS)
• Autosomal Recessive disorder characterized by
– keratitis,
– palmoplantar keratosis,
– mental retardation, and
– elevated blood tyrosine levels
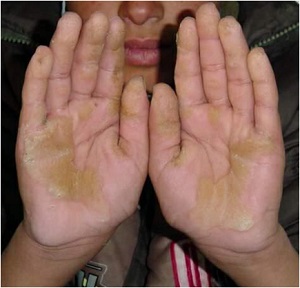
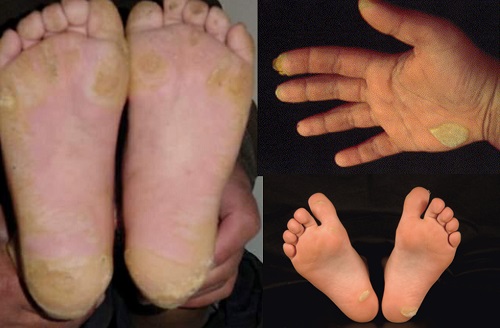
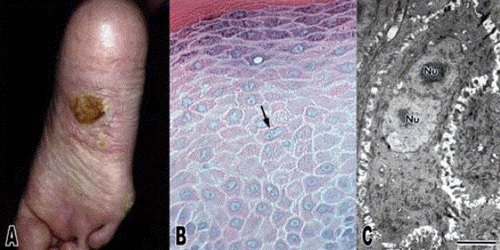
Histopathology of KERATOSIS PALMARIS ET PLANTARIS
• is nonspecific, consisting of considerable hyperkeratosis, hypergranulosis, acanthosis, and a sparse inflammatory infiltrate oflymphocyces in the upper dermis
• In epidermolytic keratosis palmaris et plantaris is identical with epidermolytic hyperkeratosis
• In keratosis palmoplantaris punctata
– massive hyperkeratosis over a sharply limited area, with depression of the underlying malpighian layer below the general level of the epidermis. increase in the thickness of the granular layer. The dermis is free of inflammation
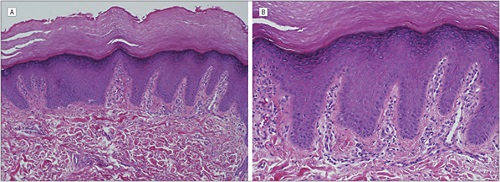
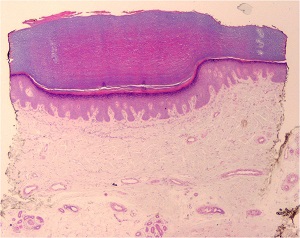
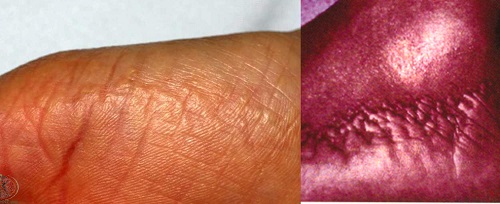
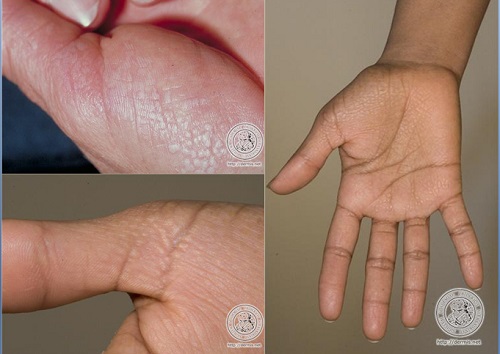
ACROKERATOELASTOIDOSIS
• Autosomal dominant
• shiny papules at the periphery of the palms and soles with extension to the dorsa of the fingers and the sides of the feet
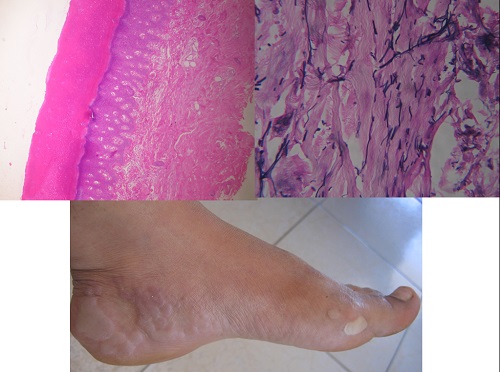
Histopathology
• Diminution and fragmentation of the elastic fibers, especially in the deeper portions of the dermis
• Some of the fragmented elastic fibers appear thickened and tortuous
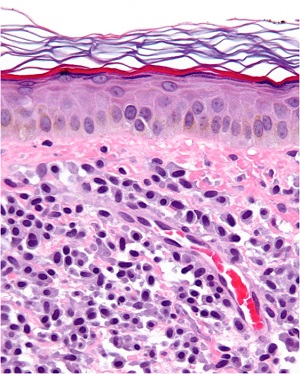
Differential Diagnosis for ACROKERATOELASTOIDOSIS
• focal acral hyperkeratosis
– the lesions have the same clinical appearance
– elastic tissue stains fail to reveal any abnormalities
• Degenerative collagenous plaques of the hands,
– late in life
– no involvement of the feet and no familial predisposition.
– histologic changes consist of basophilic degeneration of the elastic tissue
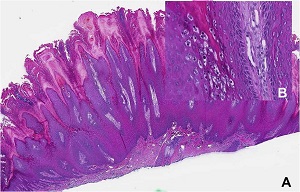
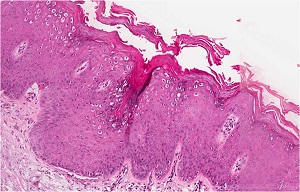
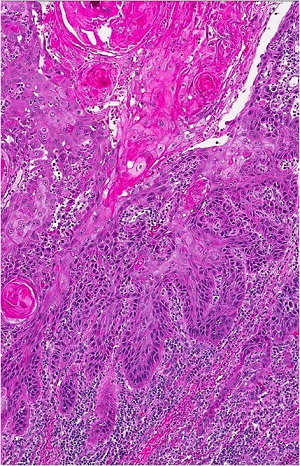
• Verruca plana lesion from chin of patient with epidermodysplasia verruciformis
• well-differentiated squamous cell carcinomas

